
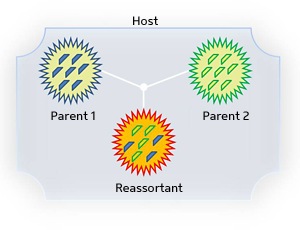
Within a single host cell, multiple influenza parent strains can swap genome segments to yield a novel hybrid ‘reassortant’ strain.
New strains of the flu emerge on a regular basis, occasionally yielding viral variants that are especially nasty and virulent. A classic example is the H1N1 ‘swine flu’ pandemic that had the world on alert in 2009. Influenza viruses evolve into new strains by undertaking reassortment—the exchange of genome segments between viruses in a host (see image). For scientists trying to determine the pedigree of a given influenza sample, understanding and sleuthing this reassortment process is a painstaking manual endeavor.
Having spent the past five years contemplating computational strategies for performing such detective work, Niranjan Nagarajan at the A*STAR Genome Institute of Singapore and Carl Kingsford at the University of Maryland, USA, have now developed a promising algorithm based on some sophisticated statistics.
“From our discussions, it was clear that there was a need for a rigorous approach with clear performance guarantees that could be used to identify flu reassortments,” says Nagarajan. He and Kingsford have developed a novel computational method, called the graph incompatibility-based reassortment finder, or GiRaF, that subjects viral genomic datasets to rigorous statistical analysis in order to derive a ‘family tree’ that accurately reflects the history of reassortment events that gave rise to that particular collection of influenza specimens.
GiRaF was shown to perform well with simulated datasets as well as a collection of 156 actual sequences from influenza H3N2, a leading cause of seasonal flu outbreaks, which had been previously subjected to manual analysis. In the case of H3N2, GiRaF successfully flagged reassortment events that had been spotted before, but also detected one more that was missed initially and revealed one previously identified event as a likely false positive. “One of the surprising results for me was the fact that we can do so well in a fully automated approach—in fact our results on real datasets were better than on our simulated datasets,” says Nagarajan. “Here was a dataset where human experts had manually analyzed the data, but missed a reassortment event found by GiRaF.”
Based on these promising results, Nagarajan believes that GiRaF should also work well for other viruses that have genomes with multiple discrete segments, as in influenza. However, he also hopes to expand the reach of the approach to other types of viruses. “Hepatitis C and dengue would be of particular interest,” he says, “as little is known about the role of recombination, if any, in viral evolution for these pathogens.”
The A*STAR-affiliated researchers contributing to this research are from the Genome Institute of Singapore.


