
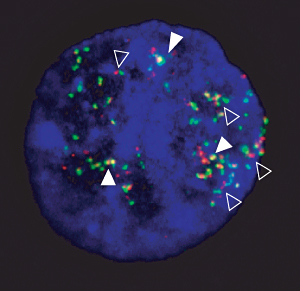
Interphase nucleus showing amplification and fusion of breakpoint flanking sequences. Double signals (filled arrowheads) indicate the fusion of the two loci and single signals indicate the normal genomic distance (open arrowheads).
© 2011 CSH Press
Cancer can leave the genome in a shambles. This is because when healthy cells become cancerous, their DNA can undergo dramatic structural rearrangements (see image), including sizeable duplications, insertions and deletions. Scientists hope to ‘zero in’ on the early events that drive tumorigenesis by mapping these changes, but existing methods have proven too expensive or time-consuming for performing large-scale analyses.
Fortunately, next-generation DNA sequencing technology that could tackle massive amounts of genomic data is now becoming accessible. Axel Hillmer and Yijun Ruan at the A*STAR Genome Institute of Singapore have recently employed such technology to map cancer-associated chromosomal alterations with unprecedented detail and accuracy.
Their method entails fragmenting chromosomal DNA into pieces of narrowly defined lengths, sequencing each fragment from both ends, and comparing the resulting genome with a ‘reference genome’ from a healthy individual. “Our technique is highly sensitive and could identify structural variations (SVs) in the genome at low sequencing costs,” says Ruan.
The researchers analyzed the genomes of normal and cancerous cell lines, including breast and stomach cancer cell lines, and assembled comprehensive maps of SVs. They found that both normal and cancerous cell lines exhibit structural rearrangements, but cancerous cells were particularly likely to accumulate certain types of SVs, such as tandem duplications, in which normally unique DNA sequences are repeated sequentially many times.
“We think tandem duplications are good candidates for triggering genomic instability,” says Hillmer. “Highly amplified rearrangement points could help to identify ‘driver’ events that have tumorigenic properties.”
Indeed, Ruan, Hillmer and their co-workers found striking evidence that certain SVs act as focal starting points for the accumulation of additional rearrangements in the same chromosomal ‘neighborhood’, suggesting a potential mechanism for cancerous transformation. However, the researchers did not observe any obvious pattern for the distribution of these driver sites. Ruan suggests that it will be necessary to examine many more samples to understand whether these specifically arise at physically fragile chromosomal sites or if these events instead emerge via disparate disruptions of common chromosome maintenance pathways.
Ruan and Hillmer are keen to further optimize the cost and speed of their technique in order to perform such analyses in the future. In addition, they believe that with further improvements, such analyses might also reach the clinic relatively soon. “We expect that within a few years our approach or similar genome sequencing methods will be applied as a routine test to determine patients’ cancer predisposition and tumor characteristics,” says Hillmer.
The A*STAR-affiliated researchers contributing to this research are from the Genome Institute of Singapore.


